

A regulatory submission can take months to prepare, but one translation issue can still slow everything down. A wrong term, an inconsistent phrase, or a missed formatting rule may lead to questions from a regulatory authority and delay approval.
That is why regulatory submission translation is not just a language task. It is part of the regulatory submission process itself.
This guide explains where submission translation usually fails, what buyers often miss, and how to build a workflow that protects your submission timeline.
Regulatory Submission Translation Has No Margin for Error
Regulatory submission translation works under strict legal requirements set by regulatory bodies in each target market. The goal is not only to make the content accurate. The goal is to make it acceptable to the regulatory agency reviewing it.
This is where many projects go wrong.
A translation can read well but still fail regulatory standards. For example, a clinical study report submitted to the European Medicines Agency must use consistent terminology across the full dossier. Product information, labeling, and information leaflets must also follow authority-specific templates and wording.
The risk becomes higher when buyers brief a provider with only the word count, languages, and deadline. Without clear regulatory requirements, the provider may not know which template, certificate, glossary, or review workflow the submission needs.
This is also where Legal Translation Services may be needed for declarations, authorizations, certificates, and legally binding documents in the submission package.
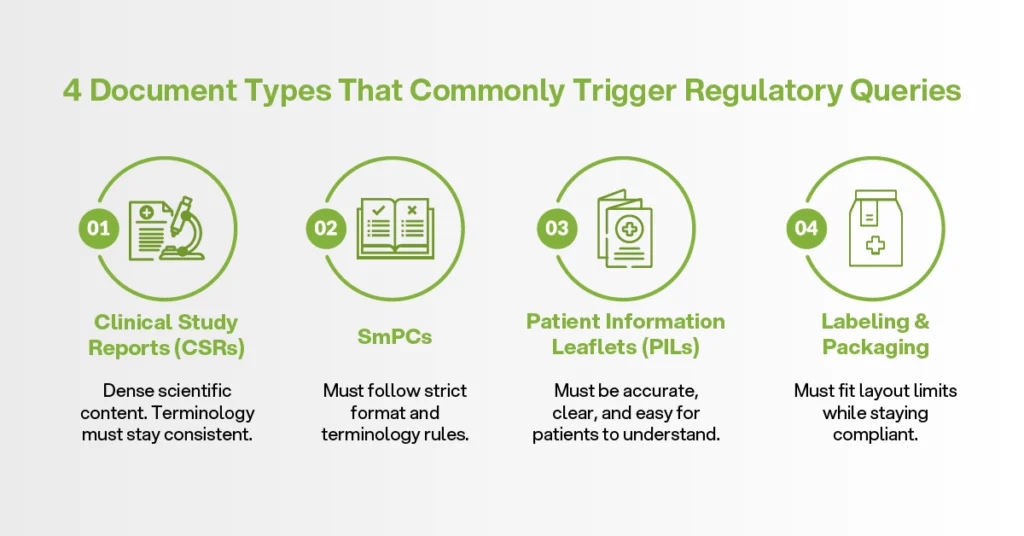
Four Document Types That Commonly Trigger Regulatory Queries
Some translation documentation carries more risk than others. In regulatory submissions, four document types need special attention.
First, clinical study reports. These are dense scientific documents, so terms related to endpoints, adverse events, dosage, and study results must stay consistent.
Second, Summaries of Product Characteristics, or SmPCs. These must follow strict format and terminology rules, especially in EU submissions.
Third, patient information leaflets. These information leaflets must be accurate, but they also need to be clear for patients. A sentence that sounds correct to a specialist may still be too difficult for the intended reader.
Fourth, labeling and packaging. Here, translation meets design limits. The wording must fit the approved layout while still meeting regulatory body requirements.
Each document type needs a different review process. Treating them all the same creates compliance gaps.
What Actually Drives Rejection Risk
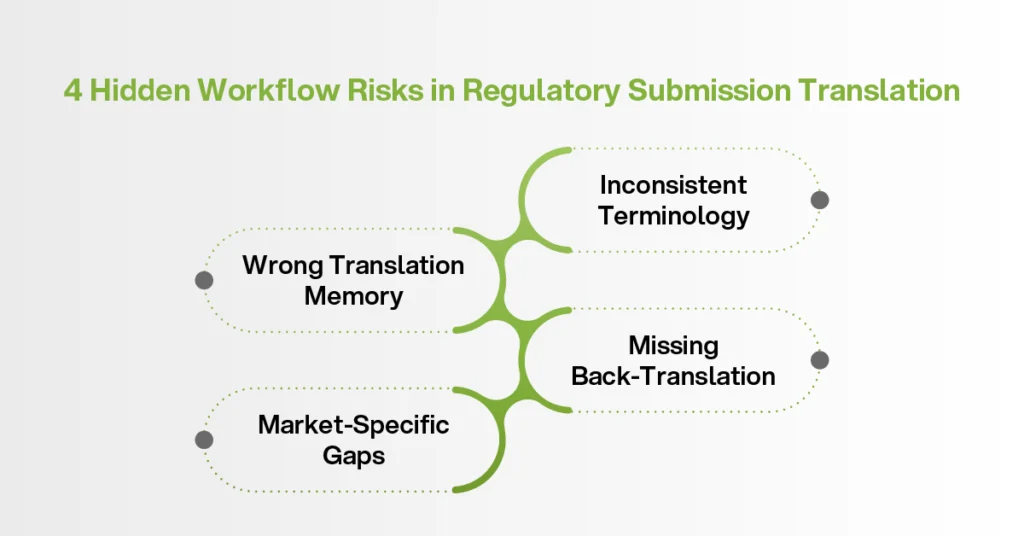
The biggest risks are often hidden in the workflow, not the language.
One common issue is inconsistent terminology across dossier modules. If different teams translate different sections without a shared glossary, the same product term may appear in different ways. To regulatory bodies, this can look like weak quality control.
Another issue is using the wrong translation memory. A general translation memory may include wording from marketing, legal, or commercial projects. That does not mean it is suitable for regulatory affairs content. Regulatory programs need a controlled translation memory built around the product, authority, and document type.
Back-translation is another missed step. Patient-facing materials, informed consent forms, and high-risk safety content may need extra validation before submission.
In multilingual regulatory submissions, these workflow gaps multiply quickly. One market may require certification. Another may require a specific format. Another may expect authority-approved terminology.
The Timeline Risk Buyers Underestimate
Translation delays affect review, formatting, QA, sign-off, and final submission.
The European Medicines Agency says the assessment of a marketing authorisation application for a new medicine can take up to 210 active days, not counting clock stops. The FDA also works with defined review goals: 10 months for standard review and 6 months for priority review.
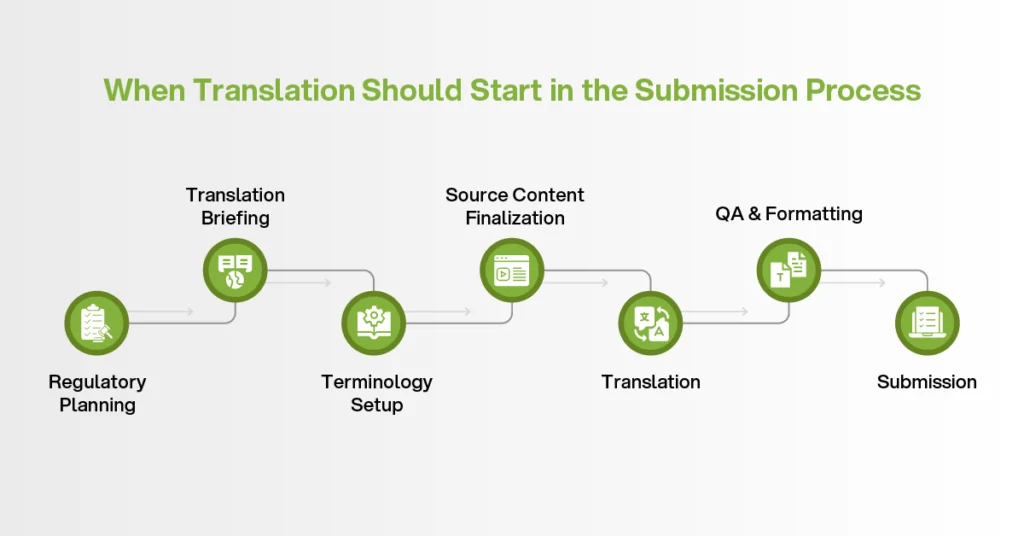
These timelines show why translation cannot be treated as a last-minute task. If translation starts only after the source files are final, the provider has less time to prepare terminology, assign specialist reviewers, load translation memory, and complete formatting checks. That pressure increases the chance of errors.
In clinical trial project management, translation planning should begin before final source delivery.
Why Not Every Language Provider Is Qualified
Not every language services provider is ready for regulatory submission work.
The difference is not only medical vocabulary. It is process control.
A qualified provider should have ISO-based workflows, experience with regulatory affairs content, specialist translators and reviewers, and documented terminology management. They should also understand the target regulatory authority.
For companies commissioning Life Sciences Translation Services, this is a key procurement point. A provider should be able to explain how they manage regulated files, who reviews them, how terminology is controlled, and how final quality is documented.
High quality is not a vague promise here. It means the translated content is accurate, consistent, traceable, and ready for regulatory use.
Step 1 — Define the Requirements Before Requesting a Quote
Before you brief any provider, define the regulatory requirements behind the project.
List the target regulatory authority, document types, language pairs, submission deadline, file formats, certification needs, notarization needs, and back-translation requirements.
For multilingual submissions, do this market by market. EMA, FDA, PMDA, and GCC requirements are not identical, so one general brief is not enough.
This step prevents scope expansion after translation has already started and gives the provider enough information to price the work properly and build the right workflow from day one.
Step 2 — Build Terminology Before Translation Begins
Terminology should not be fixed at the end. It should be controlled from the start.
Before translation begins, create three assets: a source glossary, target-language glossaries, and a dedicated translation memory for the product dossier.
The source glossary should include approved product names, active ingredients, dosage terms, safety terms, endpoint terms, and authority-specific language. The target glossaries should include validated translations for each market.
A controlled translation memory reduces repeated work across updates, labeling changes, safety reports, and future submissions. It also prevents terminology drift when large dossiers are split between several linguists.
For Pharmaceutical Translation, this infrastructure becomes more valuable over time because regulatory content is often updated, reused, and resubmitted.
Step 3 — Match the QA Workflow to the Document Type
Quality Assurance should be planned per document type, not applied as one general step at the end.
CTD modules and SmPCs may need translation, editing, proofreading, and specialist regulatory review. Patient information leaflets may need translation, editing, proofreading, back-translation, and readability review. Labeling and packaging need linguistic checks plus layout review.
This is why the QA workflow should be written into the project brief. It should include who reviews the content, what they check, which terminology assets they use, and how final approval is recorded.
Step 4 — Integrate Translation Into the Submission Timeline
The best way to reduce timeline risk is to stop treating translation as the final step.
Engage the provider four to six weeks before final source delivery. This gives the team time to review reference files, prepare glossaries, load translation memory, assign reviewers, and confirm formatting needs.
For Clinical Trial Translation Services, the same approach applies to protocols, informed consent forms, case report forms, and patient materials across multiple sites.
Translation should run as a parallel workstream beside regulatory preparation, so the final stage is cleaner and less rushed.
Regulatory Submission Translation Checklist
Before briefing a provider, confirm the following:
- Target regulatory authority for each market.
- Required document types and file formats.
- Language pairs and market-specific wording rules.
- Certification, notarization, and back-translation needs.
- Final submission deadline and internal review dates.
- Approved source glossary and reference materials.
- Dedicated translation memory for the dossier.
- QA workflow for each document type.
- Formatting and layout review requirements.
- Provider experience with regulatory affairs content and similar submissions.
Commission Your Regulatory Submission Translation Program
Regulatory submission translation needs more than accurate wording. It needs clear scope, controlled terminology, specialist review, and a workflow built around regulatory standards.
Tell bayantech your target authorities, document types, language pairs, and deadline. Our team will review your requirements and prepare a structured project scope for your regulatory submission translation program.
No unclear scope. No generic workflow. Just regulated translation support built for life sciences submissions.





